Below we’re sharing results from the validation study scientists at Diversigen and DNA Genotek undertook to ensure Diversigen’s viral metagenomics capabilities could be leveraged to help during the COVID-19 pandemic.
“When I was a boy and I would see scary things in the news, my mother would say to me, “Look for the helpers. You will always find people who are helping.”
― Fred Rogers (a.k.a. Mr Rogers)
Like we hear in the news so often, these are extraordinary (and scary) times. We’ve all had to adapt our work and home lives to stay safe and healthy during the COVID-19 global pandemic. Here at DNA Genotek, OraSure, CoreBiome, Diversigen and Novosanis, we’re working hard to ensure we can continue to offer the level of service and support you expect from us, while keeping our coworkers and loved ones safe. We’ve also watched as scientists, health care professionals, and essential service workers around the world have switched their focus from their own research, work or life goals and are asking “how can I help?” We’re doing the same.
Validation of Diversigen’s viral metagenomics sequencing and analysis pipeline for SARS-CoV-2 detection
Lisa Gamwell1 PhD, Aaron Garoutte2 PhD, Jean Macklaim3 PhD, and Emily Hollister4 PhD
1Product Manager, DNA Genotek 2. Bioinformatician, Diversigen 3. Bioinformatician, DNA Genotek 4. Director, R&D,Computational Biology, Diversigen
Diversigen has years of experience processing, sequencing and analysing samples using our proprietary viral metagenomic pipeline. Depending on the genomes of the viruses of interest, collected samples, extracted RNA or DNA, cDNA or DNA libraries and sequence data (shallow or deep, RNA or DNA based) can all be used as inputs for this pipeline. In addition to nasal nasopharyngeal samples, there are reports that SARS-CoV-2, the virus that causes COVID-19, can be detected in samples that are often of interest to microbiome researchers, including saliva and feces.
In response to the COVID-19 global pandemic, we updated the Diversigen viral database and validated our viral metagenomics pipeline to ensure they can return information about SARS-CoV-2. We determined that the pipeline can detect SARS-CoV-2 in confirmed positive samples. Using a mock community that includes 9 coronavirus strains, we determined that it can differentiate between coronavirus species. In addition, it can recover the SARS-CoV-2 genome at a depth and coverage that allows for genome assembly, which is crucial for strain and/or mutation rate tracking.
Methods and Results
To validate the viral metagenomics pipeline for the detection of SARS-CoV-2, we analyzed 5 publicly available, SARS-CoV-2 positive and 2 SARS-CoV-2 negative bronchoalveolar lavage fluid samples from The Wuhan Institute of Virology and Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College. These samples were sequenced using an RNA sequencing approach. Using our viral classification pipeline, we detected SARS-CoV-2 in all positive samples but did not detect SARS-CoV-2 in the negative samples (Table 1).
Table 1: Summary of viral classification analysis of SARS-CoV-2 positive and negative samples
Sample ID | SRA run identifier(s) | Expected SARS-CoV-2 Status | SARS-CoV-2 detected | DNA viruses detected | Other RNA viruses detected |
WIV02 | SRR11092058, SRR11092063 | ➕ | ✅ | ✅ | ✅ |
WIV04 | SRR11092057, SRR11092062 | ➕ | ✅ | ✅ | ✅ |
WIV05 | SRR11092061 | ➕ | ✅ | ✅ | ✅ |
WIV06 | SRR11092056, SRR11092060 | ➕ | ✅ | ✅ | ✅ |
WIV07 | SRR11092064, SRR11092059 | ➕ | ✅ | ✅ | ✅ |
COPD18 | SRR5677628 | ➖ | ➖ | ✅ | ✅ |
COPD25 | SRR5677642 | ➖ | ➖ | ✅ | ➖ |
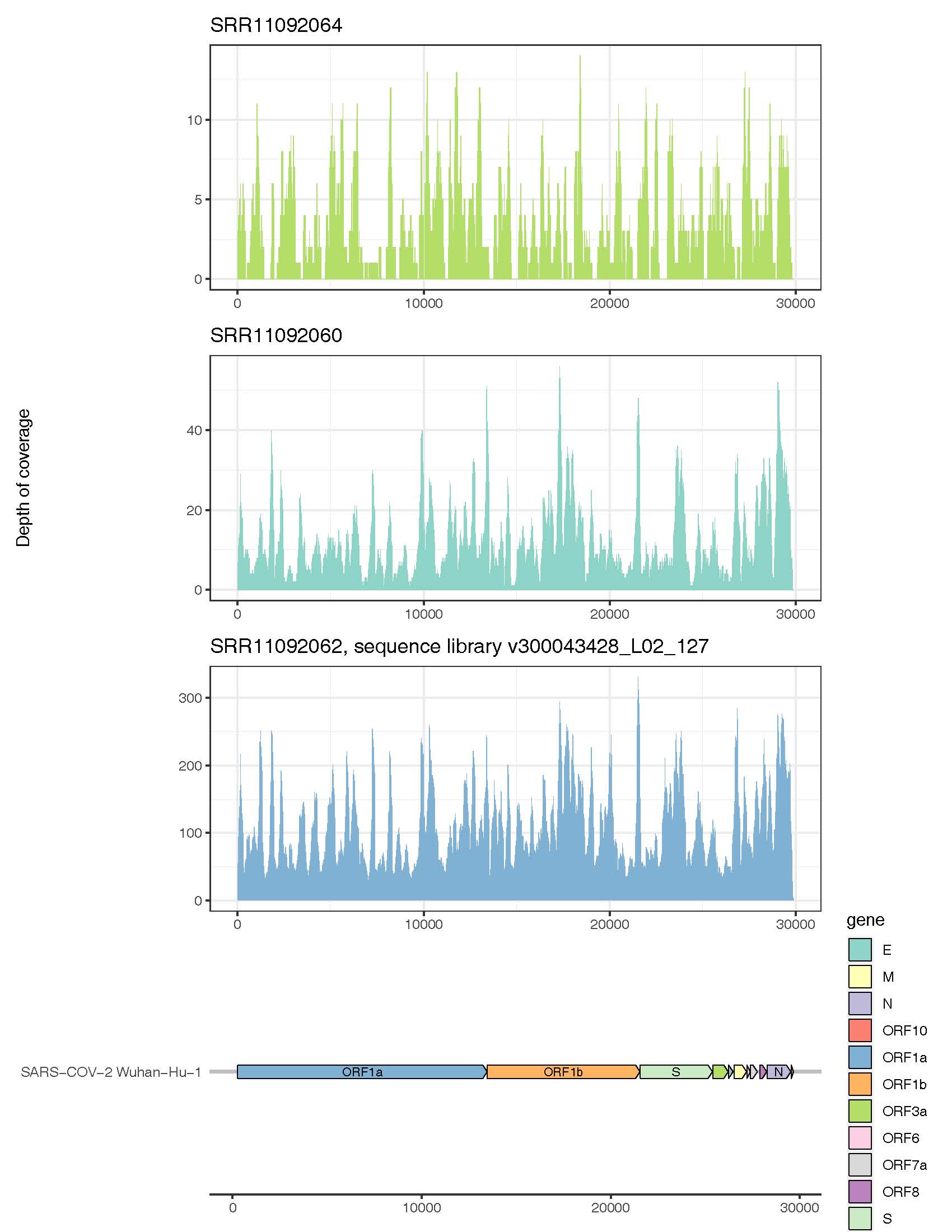
In addition to demonstrating that the SARS-CoV-2 virus could be detected in RNA sequence libraries from confirmed COVID-19 cases, we also compared recovery of the virus and its genome from sequence libraries of varying sequence depth. At depths ranging from 1-2 Gigabases (MiSeq) at the shallow end of the spectrum to 10-20 Gigabases (MGISEQ-2000RS) at the deep end, they found that they could recover 70 to 100% of the SARS-COV-2 genome with average depth of coverage ranging from 3 to >100x . The most complete recovery came from the deep sequencing samples (e.g., Table 1, SRR11092062), where we recovered a complete SARS-CoV-2 genome with average coverage of >100x. The viral metagenomics pipeline can recover and return reads sufficient for assembling high quality viral genomes. This level of coverage can facilitate strain tracking and/or mutation rate tracking, using tools like NextStrain.
Figure 1: Genome coverage of SARS-CoV-2 using Diversigen’s viral classification pipeline to analyse cDNA libraries sequenced at variable depths.
Although the occurrence of multiple coronaviruses in a single human biological specimen is likely to be rare, we created a coronavirus mock community (in silico) to evaluate the ability of our pipeline to differentiate among a variety of coronaviruses. This mock community consisted of 9 coronavirus strains, including one Alphacoronavirus, four Betacoronaviruses (including SARS-CoV-2), two Gammacoronaviruses, one Deltacoronavirus, and one unclassified Coronavirus (Table 2).
Table 2: Coronavirus Mock Community sources and descriptions
Isolate (NCBI accession) | Genome size (bp) |
Alphacoronavirus; Feline coronavirus isolate XXN (MN165107) | 29286 |
Betacoronavirus; Middle East respiratory syndrome-related coronavirus isolate llama-passaged-Qatar15 (MN507638) | 30033 |
Betacoronavirus; Dromedary camel coronavirus HKU23 isolate DcCoV-HKU23/camel/Ethiopia/CAC1019/2015 (MN514962) | 31021 |
Betacoronavirus; Middle East respiratory syndrome-related coronavirus isolate Hu/Riyadh-KSA-18013832/2018 (MN723544) | 30123 |
Betacoronavirus; Severe acute respiratory syndrome coronavirus 2 (MT121215) | 29945 |
Gammacoronavirus; Infectious bronchitis virus isolate GA/1359/199 (MN566147) | 27631 |
Gammacoronavirus; Infectious bronchitis virus isolate GA/1476/2015 (MN599049) | 27666 |
Deltacoronavirus; Porcine deltacoronavirus (MN025260) | 25399 |
Unclassified Coronavirinae; Hypsugo bat coronavirus HKU25 isolate YD131305 (KX442564) | 30498 |
Illumina reads (2×150 bp) providing 10x coverage of each genome were generated, combined, and profiled (Figure 2).
Figure 2: Expected and observed relative abundances of the Coronavirus Mock Community
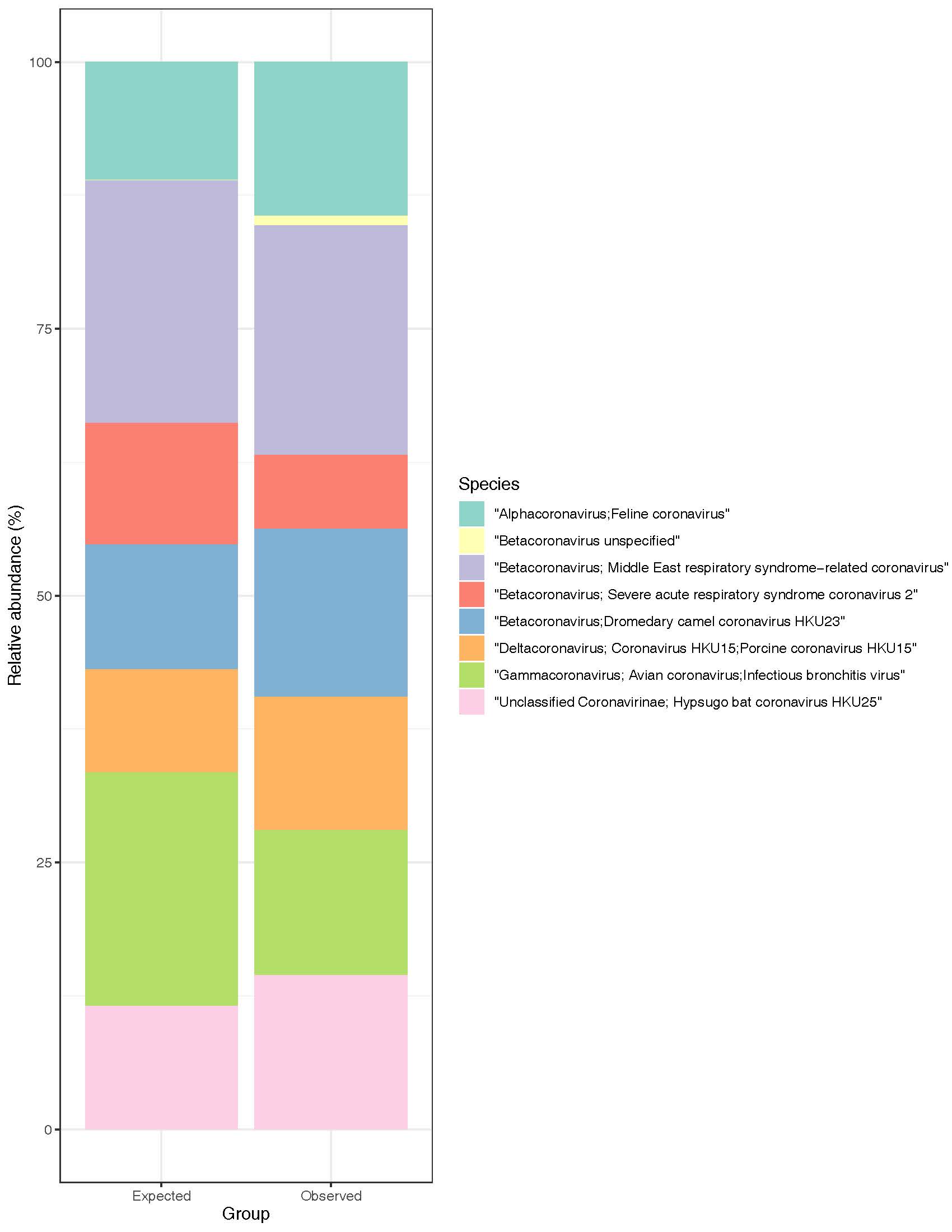
Each virus in the mock community was recovered using our analysis pipeline. It distinguished among species within the same genus (e.g., Betacoronavirus), and the majority of observed reads counts were similar to their respective expected read counts. Reads that map ambiguously (i.e., to multiple organisms in the database) are filtered during analysis, which can lead to lower than expected recovery in some cases.
Summary
Diversigen‘s viral metagenomic pipeline can not only detect SARS-CoV-2 in confirmed positive samples, it can also differentiate between coronavirus species. In addition it can be used in combination with deep sequencing to recover the SARS-CoV-2 genome at a depth and coverage that allows for genome assembly, which is crucial for tracking genetic evolution of the virus. At Diversigen, we hope we can help researchers understand how SARS-CoV-2 may be impacting their clinical trials or human research studies and support screening of donor-derived microbiome products. In addition we hope to help the research community contribute to the global efforts to track this virus.
For more information on our viral metagenomic pipeline and how we can help during the COVID-19 pandemic, please contact info@diversigen.com.
Our Viral Metagenomic Sequencing service is for research use only.
Additional Resources
Data Sharing Considerations
As always, we keep all customer data confidential, but we strongly encourage all researchers with SARS-CoV2 genetic sequence data to follow the World Health Organization (WHO) code of conduct for open and timely sharing of pathogen genetic data.
For some suggestions on where, how and why you should share SARS-CoV2 genetic sequences, please check out these links:
WHERE
The Global Initiative on Sharing all Influenza Data (GISAID),and the National Institute of Health’s genetic sequence databases, GenBank and Sequence Read Archive (SRA), host viral genomes from ongoing outbreaks, including SARS-Cov-2.
NextStrain, from the research groups of Trevor Bedford (Fred Hutchinson Cancer Research Center) and Richard Nehar (University of Basel, Max Planck Institute for Developmental Biology), provides a real-time view into the evolution and spread of a range of viral pathogens of high public health importance. Their COVID-19 specific app, Next hCoV-19 App, uses SAR-Cov2 genetic sequence data from GISAID to visualize the outbreak.
HOW
Instructions on how to upload data to the GISAID database can be found here: Submitting Data to EpiFlu™
Instructions on how to upload data to GenBank can be found here: GenBank: BankIt
WHY
Interested in learning more about how genetic epidemiology is useful during an outbreak?
Genomic Study Points to Natural Origin of COVID-19
Software and Genetic Sequencing Track the Coronavirus’s Path
NEED FUNDING? MORE INFO?
The National Institutes of Health has funding opportunities specific to COVID-19 and maintains updated information and guidance for people involved in NIH research
Coronavirus Disease 2019 (COVID-19): Information for NIH Applicants and Recipients of NIH Funding
The FDA also maintains updated information and guidance documents related to Coronavirus Disease 2019 (COVID-19), including Guidance on Conducting Clinical Trials during COVID-19 Pandemic
Do you have an important resource to share with our community? Email us and we’ll post it: info@dnagenotek.com
In this podcast, Lisa Gamwell, Services Product Manager at DNA Genotek speaks with Dan Knights, Scientific Advisor and former CEO of CoreBiome and Joy Nassif, Vice-President, Services at Diversigen, to discuss their companies and how these teams are going to work together to help microbiome researchers and the industry as a whole. Beyond that, Joy and Dan discuss some of the more unique sample types their teams have analyzed and where they see the future of microbiome analysis. You can listen to the full podcast here and selected highlights are below:
As these teams come together, how are you measuring success and ensuring you are ready to serve your customers moving forward?
“For us, success is a combined organization that continues to deliver a high level of quality science and service to our customer base. We want to be known as the preeminent microbiome support company, helping pharmaceutical and human health researchers solve problems related to the microbiome. As findings move through clinical trials, and ultimately into the clinic, we aim to be an organization capable of supporting the next phase of microbiome research in human health. That said, there is a lot of opportunity outside of human health, whether it’s animal health, agriculture or environmental, they are burgeoning with respect to microbiome and give us areas to grow moving forward.”
Joy Nassif
Both teams have processed some unique samples (ex. moon rocks, make-up, wine). What are some challenges in analysing unique sample types?
“You have challenges on the physical wet-lab side, and you have challenges on the data/informatics side. Physically, the goal is to get plenty of microbial DNA out of a sample so that you can sequence, quantify and explore it. If you have a tricky material, such as cotton, it can be hard to get the microbial DNA out, either because it has low amount of microbial DNA or because it has a ton of other DNA contaminating the sample. Once you solve the wet-lab problems and get the DNA out, now you’re in the virtual world and need to analyze it. You have to bring different methods to bear on samples where the microbes are totally unknown and not found in the standard databases.”
Dan Knights
What are some challenges in processing human samples, such as stool?
“We love stool samples! The signal is strong and they are very robust. There can be challenges with samples from patients with certain diseases, those who have undergone chemotherapy or people who have Clostridium difficile infections, as these patients can have very low amounts of bacterial DNA and can have more host DNA contamination. However, for the most part, the gut microbiome is the easiest to work with.”
Dan Knights
“How you are collecting your samples ultimately determines how successful you are going to be through anyone’s pipeline in the wet-lab. We’ve always been fans of DNA Genotek’s stool collection kit, for stability and compatibility reasons. When you put garbage in, you get garbage out – but when you have a high quality collection device, your method of collection makes sense, and you’re working with an experienced microbiome laboratory, you should have success.”
Joy Nassif
As the industry moves towards multiomics, what “omics” do you see helping provide answers in the future? And what are the challenges?
“The first omic of interest to many of our customers today is metabolomics. I think that will be a primary output that researchers would like to start seeing integrated with microbiome data. Human genomic data, as it relates to specific disease and how it interacts with the microbiome data, will be important as well.”
Joy Nassif
“The challenge in combining metabolomics and microbiome analysis or genomic analysis, is integrating the structures that we already know about into the analysis. Taking advantage of all of the prior knowledge we have about what metabolic pathways exist, what are the inputs and outputs, what genes are in those pathways. That provides us the framework to leverage the data better than if we just treated every metabolite or microbe as an arbitrary feature.”
Dan Knights
Want to learn more about our end-to-end microbiome solution? Reach out to discuss how Diversigen can help with your project.
